Le repiquage cellulaire, également désigné par les termes de passage ou de subculture, représente une étape cruciale et récurrente en culture cellulaire. Il s'agit d'un processus essentiel pour maintenir la viabilité et la prolifération des cellules in vitro lorsque celles-ci atteignent une densité critique, appelée confluence. Lors des repiquages ou passages, quand les cellules arrivent à confluence, on dissocie les cellules pour pouvoir les diluer dans un milieu frais. Cette procédure permet non seulement de prévenir l'inhibition de contact et l'épuisement des nutriments, mais aussi de générer de nouvelles cultures pour des expérimentations ultérieures ou pour le stockage.
Les Principes Fondamentaux du Repiquage Cellulaire
Le principe central du repiquage consiste à prélever une fraction de cellules d'une culture établie, à les dissocier si elles sont adhérentes, puis à les ressemer dans de nouveaux récipients de culture avec un milieu frais et une densité appropriée. Lorsque les cellules deviennent confluentes, leur prolifération ralentit en raison du contact cellulaire et du manque d'espace. Pour maintenir leur état physiologique et leur capacité de division, on procède à une dissociation mécanique et/ou enzymatique, suivie d'un comptage et d'un repiquage dans de nouveaux flacons à une densité adaptée. Il est préférable de repiquer à une confluence entre 70 et 90%.
L'Importance de la Confluence et de l'Adhérence Cellulaire
Le niveau de confluence, c'est-à-dire le pourcentage de la surface couvert par les cellules, est un paramètre clé pour décider du moment du repiquage. Pour les cellules qui prolifèrent, on suit le niveau de confluence. Un repiquage trop tardif peut entraîner un vieillissement prématuré des cellules, une perte de leurs caractéristiques physiologiques ou même leur mort. Inversement, un repiquage trop précoce peut ne pas justifier l'effort et le coût, et peut potentiellement diluer une culture trop faible.
Les cellules sont cultivées sur du plastique traité pour qu'elles puissent y adhérer ou sur des éléments de matrice extracellulaire que l'on dépose avant d'ensemencer les cellules (collagène, fibronectine ou encore le Matrigel (lame basale produite par des cellules de sarcome de souris) qui par son épaisseur permet de créer un environnement 3D). Ces substrats facilitent l'attachement des cellules, un processus essentiel pour la plupart des lignées cellulaires adhérentes. Des techniques avancées, telles que la technologie xCELLigence, permettent un suivi en temps réel de l’adhérence et de la confluence des cellules. Cette technologie est basée sur les variations de l’impédance (de la résistance électrique) d’une couche riche en or qui dépend des cellules qui sont attachées sur le substrat. Ces modifications d’impédance sont traduites en un « cell index » qui est nul si aucune cellule n’est attachée. Des changements dans la morphologie cellulaire (adhérence, étalement), le nombre de cellules (prolifération ou mort) ou la migration peuvent être étudiés à l’aide de ce système. En haut, on suit l’adhérence initiale de cellules que l’on vient de disperser dans le milieu de culture. En bas, à plus long terme, on peut suivre l’étalement des cellules. Ici, les cellules ne prolifèrent pas, mais si elles proliféraient on observerait une pente ascendante au lieu d’un plateau.

Les Méthodes de Dissociation Cellulaire
La dissociation des cellules est une étape critique du repiquage, particulièrement pour les cultures adhérentes. Cette étape vise à détacher les cellules de leur substrat et les unes des autres sans compromettre leur viabilité.
La Dissociation Enzymatique : La Trypsine et l'EDTA
La méthode la plus courante pour dissocier les cellules est l'utilisation d'enzymes protéolytiques. La trypsine est l'enzyme la plus fréquemment utilisée à cette fin. La trypsine est disponible sous forme d’extrait brut de pancréas de porc en solution. Elle agit en digérant les protéines responsables de l'adhérence cellulaire à la matrice extracellulaire.
L'EDTA (acide éthylènediaminetétraacétique) est souvent combiné à la trypsine. L'EDTA piège les ions Ca2+ nécessaires aux cadhérines qui permettent l’adhérence des cellules entre elles. Les lignées dérivant d’épithélium peuvent exprimer aussi des cadhérines connues pour créer des jonctions adhérentes fortes calcium-dépendantes appelées desmosomes. Dans ce cas, l’ajout d’un chélateur du calcium comme l’EDTA peut être nécessaire pour les dissocier. Le calcium et le magnésium présents dans la matrice extracellulaire aident l’adhésion entre cellules en protégeant les liaisons peptidiques sur lesquelles agit la trypsine.
Protocoles d'Utilisation de la Trypsine-EDTA :
Étant donné qu’il n’y a pas de protocole universel, la nature des cellules et le milieu de culture sont des paramètres importants à prendre en compte pour déterminer le meilleur protocole.
- Préparation : Le sérum contient naturellement un inhibiteur de la trypsine (alpha2-macroglobuline et des facteurs d’attachement/adhérence cellulaire (fibronectine)). Le sérum étant un inhibiteur de la trypsine, il faut également laver les cellules avec du PBS avant la trypsinisation pour éliminer toute trace de sérum.
- Application de la Trypsine : La trypsine concentrée (10X) est fournie à 25 g/l soit 2.5%. Il convient d'ajouter une quantité appropriée de solution de trypsine-EDTA pour couvrir le fond du flacon :
- Dans une flasque 25 cm², ajouter environ 0,5 à 1 ml.
- Dans une flasque 75 cm², ajouter environ 2 à 3 ml.
- Dans une flasque 175 cm², ajouter environ 5 à 7 ml.
- Laisser agir le film de trypsine qui subsiste sur les cellules pendant quelques minutes.
- Surveillance : Au bout de 5 minutes, vérifier régulièrement le décollement des cellules (les cellules s’arrondissent). Cela peut être observé au microscope.
- Inactivation : Quand on rajoute sur les cellules du milieu frais contenant du sérum, on bloque naturellement l’action de la trypsine. Ce n’est pas le cas d’une culture sans sérum où l’on doit bloquer artificiellement son activité.
- Récupération des Cellules : Une fois les cellules décollées, on les resuspend dans un milieu de culture frais, souvent additionné de sérum pour neutraliser l'activité résiduelle de la trypsine.
Alternatives à la Trypsine : L'Accutase et Autres Enzymes
Dans le cas d’une culture en milieu sans sérum ou de cellules sensibles (cellules primaires et cellules souches), il est indispensable d’adoucir l’étape de trypsinisation et de contrôler les conditions pour maintenir une bonne viabilité des cellules.
L’Accutase® est un cocktail d’enzymes qui fonctionne comme la trypsine, mais a l’avantage de s’autodétruire. En effet, sa température optimale pour fonctionner est de 25°C. Son activité diminue à 37°C et devient inactive en quelques minutes à 37°C. Cela offre un avantage significatif pour les cellules sensibles, réduisant le risque de dommages enzymatiques prolongés.
D'autres enzymes peuvent être utilisées pour la dissociation, notamment la dispase et la collagénase, qui sont souvent employées pour des tissus plus complexes ou pour dissocier des cellules nécessitant une approche plus douce. Augmenter la concentration d’enzyme, d’EDTA ou ajouter d’autres enzymes (ex : dispase, collagénase) peut être nécessaire pour certaines lignées.
Germ Cells Purification by Tissue Dissociation and Flow Cytometry | Protocol Preview
Les Problèmes Courants et Leurs Solutions lors du Repiquage
Le repiquage cellulaire, bien que courant, peut être confronté à plusieurs défis qui affectent la viabilité et la prolifération des cellules.
Problèmes d'Adhérence Post-Repiquage
Une mauvaise adhérence des cellules après le repiquage est un problème fréquent. Plusieurs facteurs peuvent en être la cause :
- Dissociation excessive : L’étape de dissociation a été excessive et des cellules lysées ont relargué de l’ADN génomique. Pour y remédier, ajouter une goutte de DNase stérile (1 mg/ml dans l’eau) aux cellules en suspension pour casser l’ADN.
- Dommages Cellulaires : Les cellules ont été centrifugées trop brusquement ou trop longtemps au moment de retirer l’excès de solution de dissociation, ou l’étape de dissociation a été trop longue et a endommagé des protéines membranaires nécessaires à l’adhérence.
- Milieu de Culture Inadapté : Une concentration insuffisante de sérum ou de facteurs d’attachement dans le milieu de culture (courant avec les milieux sans sérum) peut compromettre l'adhérence. Le milieu ne convient pas.
- Inactivation Incomplète : La solution de dissociation n’a pas été inactivée ou retirée par centrifugation, laissant des enzymes résiduelles endommager les cellules.
- Conditions de Stockage Inappropriées : Les cellules détachées en suspension ont été laissées trop longtemps avant de réensemencer.
- Paramètres Physico-chimiques Incorrects : Le pH ou l’osmolarité de la solution saline tamponnée est incorrecte.
Vieillissement Cellulaire et Sénescence
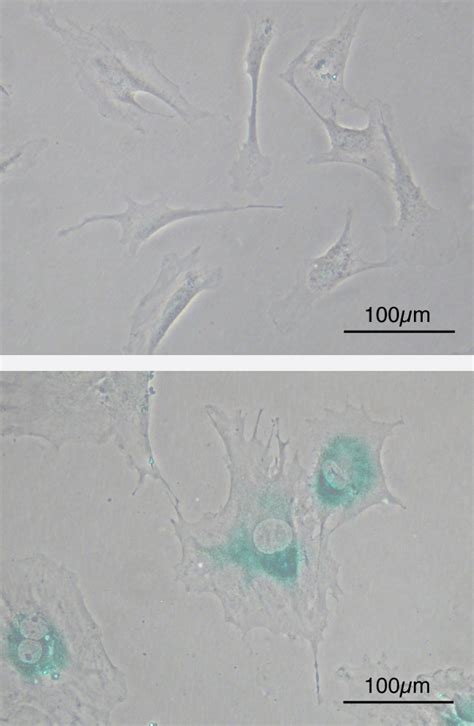
La sénescence cellulaire est un état d'arrêt permanent du cycle cellulaire qui peut être induit par plusieurs facteurs, y compris les repiquages répétés. Dans des cultures de fibroblastes de souris embryonnaires (MEF), on observe avant la sénescence, au 2ème passage après prélèvement, des MEF ayant une forme de fuseau. Après le 8ème passage, les MEF se sont agrandis et se sont aplatis. La coloration bleu-vert indique l’expression de la β-galactosidase associée à la sénescence (la coloration est obtenue en présence du substrat X-gal). Les lignées de cellules cancéreuses ne connaissent pas ces phénomènes de sénescence. Les cellules HeLa, par exemple, isolées en 1951, ont constitué la première lignée cellulaire humaine immortelle et continuent d'être un outil fondamental en biologie cellulaire.
Le Contexte Plus Large de la Culture Cellulaire
La culture cellulaire présente de nombreux avantages mais aussi des inconvénients. Les cellules sont dans un environnement contrôlé (température, nutriments disponibles, éventuellement en présence de matrice extracellulaire…) mais cet environnement est parfois éloigné des conditions physiologiques. Dans des cultures classiques en 2D chaque cellule est facilement visible, mais les propriétés des organisations 3D dans les tissus peuvent être perdues.
Applications de la Culture Cellulaire au-delà du Repiquage
Le repiquage cellulaire est une technique de base qui rend possibles des applications plus complexes en biologie cellulaire et moléculaire.
Le Fractionnement Cellulaire
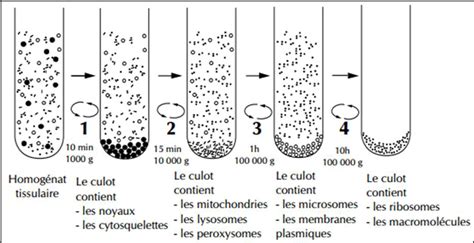
Au-delà du simple maintien des cultures, les techniques de repiquage sont souvent combinées à des méthodes de fractionnement cellulaire. Il s’agit d’une technique pour purifier des fractions particulières du contenu des cellules (noyau, mitochondries, ribosomes…). L'étude des fractions protéiques cytoplasmiques et nucléaires dans un modèle de maladie de Huntington en est un exemple. On soumet des neurones avec des allèles sauvages (+) ou mutants (109) pour le gène codant la huntingtine à un fractionnement cellulaire par centrifugation qui permet de récupérer les noyaux dans le culot et le cytoplasme qui se retrouve dans le surnageant. On réalise des extraits protéiques de ces deux fractions que l’on analyse ensuite par western-blots avec des anticorps reconnaissant la protéine REST/NRSF ainsi que des anticorps reconnaissant la tubuline (contrôle des fractions cytoplasmiques) et Histone H1 (contrôle des fractions nucléaires). On constate que dans les neurones mutants, REST/NRSF a une localisation nettement plus nucléaire que dans les neurones sauvages au détriment de la localisation cytoplasmique. Le fractionnement a été correctement réalisé car on ne retrouve pas de tubuline dans les fractions nucléaires et pas d’Histone H1 dans les fractions cytoplasmiques.
Le Western-blot : Analyse des Protéines
Le Western-blot est une technique couramment utilisée pour détecter et quantifier des protéines spécifiques dans des extraits cellulaires. Les cellules ont été lysées dans du tampon RIPA (150 mM NaCl, 1 % NP-40, 0,25 % Na-désoxycholate, 50 mM Tris-HCl [pH 7,4]) contenant 1 comprimé d’inhibiteur de protéase et 1 comprimé d’inhibiteur de phosphatase par 10 ml. La concentration totale de protéines dans les lysats a été déterminée à l’aide d’un kit Pierce BCA Protein Assay. L’absorbance a été mesurée à l’aide d’un spectrophotomètre à microplaque.
Un gel de polyacrylamide à 8 % contenant du SDS surmonté d’un gel de stacking (pour concentrer les protéines) a été préparé. Ensuite, 15 µL d’échantillon, contenant 9 µg de protéines totales et 5 µL de tampon d’échantillon (5,7 ml d’eau, 1,6 ml de glycérol (densité importante pour que l’échantillon aille au fond du puits), 1,1 ml de SDS à 10 % (dénaturation), 1,3 ml de Tris 0,5 M (pH 6,8), 25 mg de dithiotréitol (DTT, casse les ponts disulfures des protéines), 300 µL de bleu de bromophénol (pour bien visualiser l’échantillon)) ont été chauffés à 90 °C pendant 5 min (pour dénaturer les protéines), refroidis sur de la glace et chargé sur le gel.
Le gel a été incubé dans du tampon d’électrophorèse (Tris Base 25 mM, glycine 190 mM, 0,1 % SDS) à 70 V jusqu’à ce que les échantillons atteignent le gel de séparation, puis à 150 V jusqu’à ce que les échantillons atteignent le bas du gel. Ensuite, les protéines ont été transférées sur une membrane en polyfluorure de vinylidène (PVDF) pendant 1 h à 80 V sur glace dans un tampon de transfert froid (Tris base 25 mM, glycine 190 mM, éthanol 20%). La membrane a été rincée à l’eau et lavée 2x dans une solution saline tamponnée au Tris et du Tween-20 (TBS-T) (20 mM de Tris/HCl, 137 mM de NaCl, 0,1 % de Tween-20).
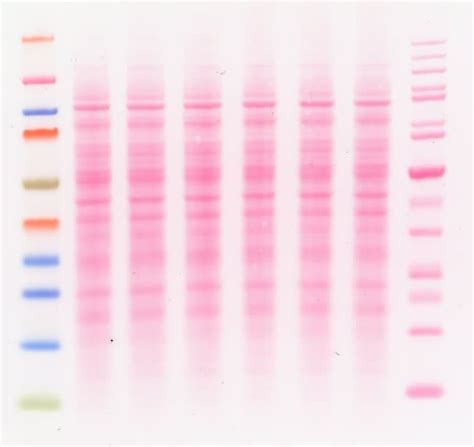
La membrane a été incubée pendant 1 h à température ambiante dans un tampon de blocage (5% BSA dans du TBS-T, sature les sites non spécifiques sur lesquels l’anticorps pourrait s’accrocher) sous agitation. Ensuite, la membrane a été incubée pendant la nuit dans le tampon de blocage avec un anticorps primaire à la concentration souhaitée à 4°C sous agitation. La membrane a été lavée 3 × 5 min dans du TBS-T et incubée dans le tampon de blocage avec l’anticorps secondaire reconnaissant l’anticorps primaire pendant 1 h à température ambiante. Un ECL a été réalisé. Une membrane de western-blot colorée au Rouge Ponceau permet de contrôler si le transfert s’est bien passé et si la quantité totale des protéines est similaire d’un puits à l’autre (ici, il y a un peu moins de protéines dans le puits à gauche).
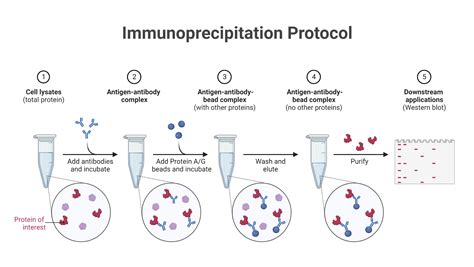
L'Immunoprécipitation et la Détection des Interactions Protéiques
L'immunoprécipitation (IP) est une technique puissante pour isoler une protéine d’un extrait cellulaire et tous les interactants directs ou indirects avec cette protéine grâce à l’interaction très spécifique anticorps-antigène. Cette technique permet de savoir si deux protéines A et B appartiennent à un même complexe à un moment donné et dans des conditions données. On fait une immunoprécipitation avec un anticorps contre A et on analyse la présence par western-blot de la protéine B reconnue par un autre anticorps. Les anticorps peuvent être fixés sur des billes et on peut récupérer dans le culot les complexes formés après une centrifugation. Alternativement, on peut utiliser des billes métalliques qui sont ensuite attirées par un aimant.
Un exemple d’immunoprécipitation : la mise en évidence de la présence dans un même complexe des protéines BRCA1 et ER-α. Des extraits protéiques de cellules de tumeurs de glande mammaire MCF-7 (qui expriment beaucoup de récepteurs aux œstrogènes) et de tumeurs de prostate Du-145 (qui expriment très peu de récepteurs aux œstrogènes) sont soumis à une immunoprécipitation (IP) avec un anticorps anti-BRCA1 ou anti-ER-α. Une immunoprécipitation avec un anticorps Ig est réalisée à titre de témoin négatif. Ces fractions immunoprécipitées sont soumises à un western-blot avec un anticorps anti-BRCA1 ou anti-ER-α. On vérifie d’abord qu’il y a bien du ER-α et du BRCA1 dans la fraction précipitée avec leurs propres anticorps respectifs. On constate ensuite la présence de ER-α dans la fraction précipitée avec l’anticorps anti-BRCA1 et la présence de BRCA1 dans la fraction précipitée avec l’anticorps anti-ER-α, seulement dans les cellules MCF-7.
Les "tags" sont de courtes séquences d'acides aminés ajoutées à une protéine d'intérêt pour faciliter sa détection ou sa purification. Séquence de quelques tags communément utilisés : HA, Myc, Flag, GST, MBP. L'interaction entre Myc-MT1X et HA-FHL3 peut être démontrée par immunoprécipitation même si l'on ne dispose pas de bons anticorps reconnaissant spécifiquement la protéine MT1X et FHL3. Des cellules 293T ont été transfectées avec des plasmides permettant d’exprimer des protéines MT1X couplées au tag Myc et FHL3 couplées au tag HA. Des extraits protéiques sont réalisés à partir de ces cellules et traitées en western-blot avec des anticorps anti-HA et anti-Myc (input). On réalise ensuite une immunoprécipitation (IP) avec un anticorps IgG (témoin négatif) et un anticorps anti-HA (ce qui revient à immunoprécipiter HA-FHL3) et on réalise ensuite une analyse par western-blot du culot obtenu avec un anticorps anti-HA (on vérifie que l’immunoprécipitation a bien fonctionné) et avec un anticorps anti-Myc (on cherche à savoir si Myc-MT1X se trouve dans le culot avec HA-FHL3 montrant que FHL3 et MT1X sont présents dans un même complexe).
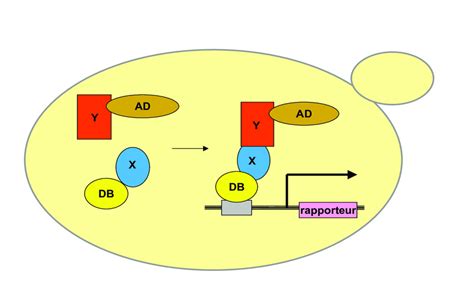
Le Système du Double Hybride
Le système du double hybride effectué chez la levure ou dans des cellules de mammifères permet de mettre en évidence l’interaction entre deux protéines. Il est basé sur la nature modulaire des facteurs de transcription qui sont généralement composés de deux domaines : un domaine de liaison à l’ADN et un domaine d’activation de la transcription. Chez la levure, on utilise le facteur de transcription Gal4. Si on veut savoir si la protéine A interagit avec la protéine B, on fait synthétiser aux cellules deux protéines de fusion : une avec le domaine de liaison à l’ADN de Gal4 fusionné avec la protéine A et une autre avec le domaine d’activation de Gal4 fusionné avec la protéine B. Les protéines fusion A-Gal4DBD (DBD = domaine de fixation à l’ADN) et B-Gal4AD (AD = domaine d’activation de la transcription) doivent interagir pour que la transcription du gène rapporteur (en orange) puisse être activée. Le domaine de fixation à l’ADN de Gal4 se fixe sur une séquence appelée UAS.
Un exemple d’analyse en double-hybride avec la levure montre la croissance de colonies de levure sur milieu avec histidine (+His) ou sans histidine (-His). Les levures sont déficientes en l’enzyme HIS3 qui permet de synthétiser l’histidine qui est indispensable pour la croissance, mais elles possèdent une construction où le gène codant cette enzyme est sous le contrôle d’une séquence UAS. Les levures exprimant la protéine DDB1 fusionnée au domaine de liaison à l’ADN de GAL4 (BD-DDB1) sont co-transformées soit avec le vecteur AD vide (juste le domaine d’activation de la transcription de GAL4), soit avec les constructions AD-CUL4 ou AD-DET1 où ce domaine est fusionné avec les protéines CUL4 ou DET1, respectivement. Différentes doses de plasmides avec les différentes constructions AD sont testées (gradients horizontaux en haut). En présence d’histidine (+His), toutes les souches poussent, montrant qu’il n’y a pas de toxicité liée à la transformation. En absence d’histidine (−His), seule la co-expression BD-DDB1/AD-CUL4 et BD-DDB1/AD-DET1 permet la croissance, indiquant l’activation du gène rapporteur HIS3 par interaction spécifique entre DDB1 et CUL4 ou DET1. On remarque un effet dose plus marqué pour AD-DET1 montrant que l’interaction entre DDB1 et DET1 est plus fragile qu’entre DDB1 et CUL4.
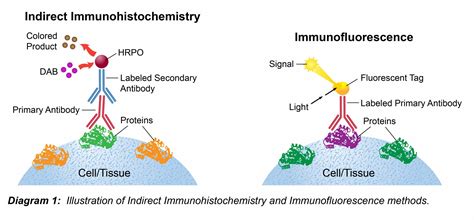
Immunohistochimie et Immunofluorescence
Comme le western-blot, ces méthodes utilisent la spécificité de reconnaissance des anticorps pour mettre en évidence les protéines d’intérêt mais cette fois-ci non pas à partir d’extraits protéiques mais dans des cellules (cyto) ou des tissus (histo) qui ont été fixés au préalable.
Un exemple d’immunohistochimie sur un embryon de poulet permet de reconnaître les cellules de crêtes neurales (anticorps primaire HNK-1 (en vert) qui reconnaît un antigène à la surface de ces cellules). La fixation est réalisée avec du paraformaldéhyde (PFA) 4% qui donne du formaldéhyde en solution aqueuse. Il permet de préserver les structures cellulaires en pontant les protéines entre elles. On doit réaliser une perméabilisation pour la bonne pénétration des anticorps avec des détergents non ioniques comme le Tween 20 ou le Triton X-100. L’anticorps primaire (vert) reconnaît spécifiquement l’antigène (triangle vert). L’anticorps secondaire (rouge) reconnaît le premier anticorps (il doit être choisi pour reconnaître les anticorps de l’espèce dans laquelle a été produit l’anticorps primaire) et il est couplé à la peroxydase qui, en présence de DAB (3,3′-diaminobenzidine) et d’H202, donne un produit coloré brun. L'immunohistochimie montre que les cellules de crêtes neurales ne migrent que dans la moitié antérieure des somites. Vue latérale de la région troncale d’un embryon de poulet immunomarqué avec l’anticorps HNK-1 qui reconnait les cellules de crêtes neurales. La partie dorsale est vers le bas. On constate que les cellules de crête neurale, bien qu’elles sortent uniformément le long de l’axe antéro-postérieur du tube neural, migrent uniquement dans la partie antérieure des somites.
L’immunochimie est basée sur la révélation d’une activité enzymatique apportée par l’anticorps secondaire, alors que l’immunofluorescence est basée sur l’émission de photons à une longueur d’onde donnée d’un fluorophore couplé à l’anticorps secondaire. L’anticorps primaire dirigé contre la protéine d’intérêt X est reconnu ensuite par un second anticorps dirigé contre l’anticorps primaire et qui est couplé à un fluorophore.
tags: #repiquage #cellulaire #def